
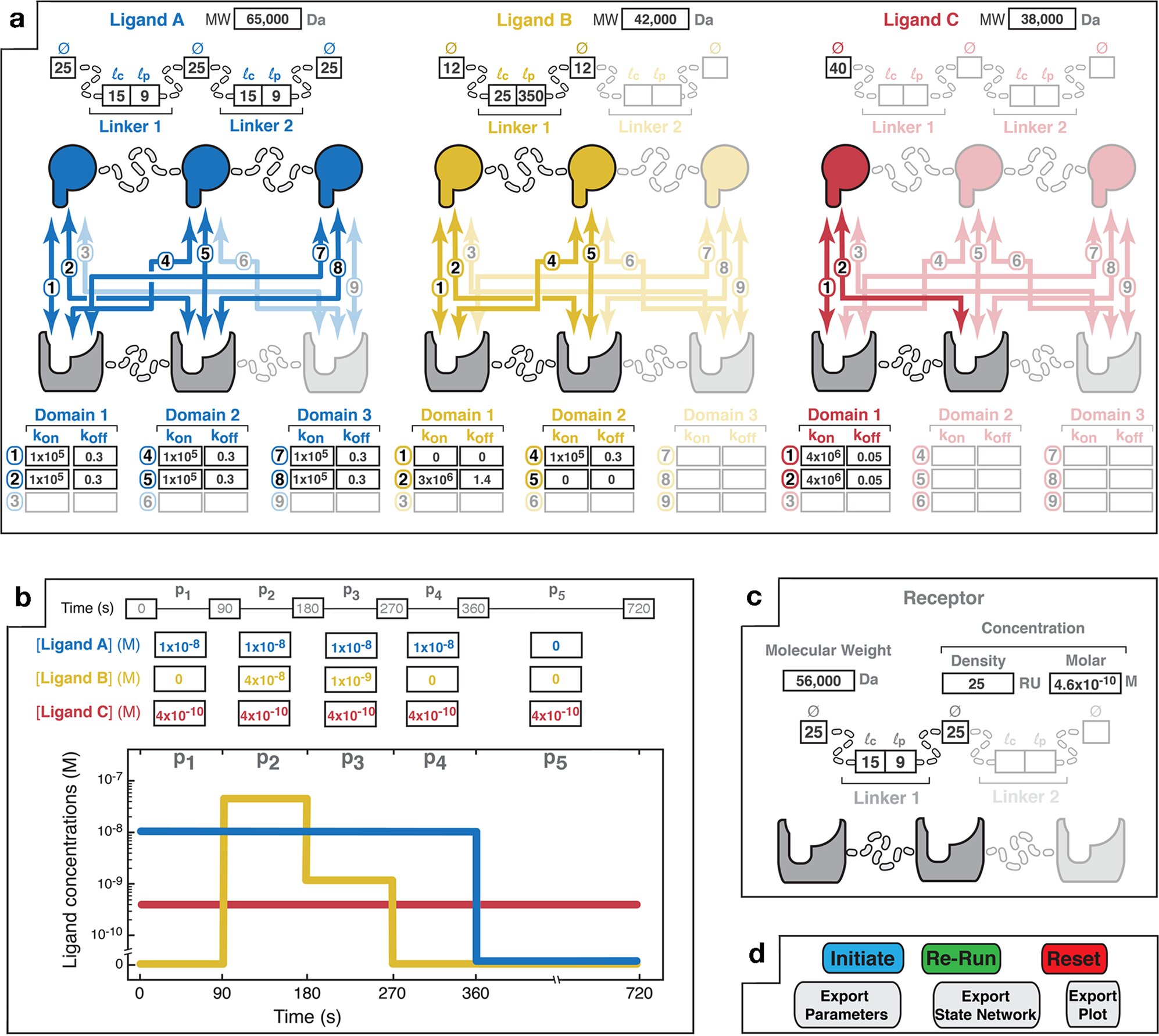
{kind=link}
 and valencies of the ligand(s) and receptor (up to trivalent) that compose the multivalent system. Based upon the chosen design, the user specifies the structure of each of the ligands by entering the applicable molecular weight (MW); the binding domain diameters (Ø); the contour lengths (lc of the linkers (i.e., the maximum end-to-end distance; e.g., 3.5 Å and 1.5 Å per amino acid for a random coil and alpha helix, respectively); and the persistence lengths (lp) of the linkers. Further, the applicable combinatorial interactions (numbered 1 to 9) unique to each receptor–ligand pairing are highlighted. Parameter fields allow the input of monovalent rate constants for each pairwise interaction. Non-binding interactions can be indicated with <i>k</i><sub>on</sub> and <i>k</i><sub>off</sub> values of zero (e.g., as illustrated with Ligand B in yellow for interactions \"1\" and \"5\"). <b>b</b> An input field allows the user to specify patterns of the total, bulk ligand concentrations. An association phase occurs during periods of non-zero bulk ligand concentration (e.g., 90–270 s for Ligand B). Dissociation phases occur when the ligand is removed from the bulk solution (e.g., 360–720 s for Ligand A). Here, Ligand C is specified as continuously present in solution during the 720 s of the interaction timecourse. The graphical display allows visualization of the specified bulk concentration pulse pattern. <b>c</b> User input parameters for the receptor. Receptor concentration can be specified as either an SPR-mimicking surface density (measured in RU; where 1 RU equals ~1 pg/mm<sup>2</sup>) or a molar concentration. Receptor topology is specified in the same form as described above for the ligands. <b>d</b> The <i>MVsim</i> controller tab enables initiation, iteration, and export of binding simulations. \"Initiate\" executes a simulation. \"Re-run\" executes an abbreviated simulation used when no changes were made to the valency or topology of the system. \"Reset\" relaunches the app and clears user input parameters from all fields. Credit: <i>Nature Communications</i> (2022). DOI: 10.1038/s41467-022-32496-6")
A crew led by College of Minnesota Twin Cities biomedical engineers has developed a universally accessible software that may simulate complicated molecular interactions, which is able to permit researchers to design higher therapies for ailments like most cancers and COVID-19.
The paper builds upon a research the researchers revealed in 2019. Now, they’ve expanded the expertise to simulate much more complicated molecular interactions, made the applying straightforward for non-experts to make use of, and utilized their findings to make clear how the SARS-CoV-2 virus infects the physique.
The research is revealed in Nature Communications, and the app, referred to as MVsim, is freely obtainable to different researchers on GitHub.
The simulator predicts the energy, pace, and selectivity of multivalent interactions, which contain molecules which have a number of binding websites and can be utilized to develop medicines for ailments, notably most cancers and COVID-19.
“Multivalent interactions are actually essential in pure organic techniques, and they’re now beginning to be creatively exploited for creating new therapeutic medication that leverage their distinctive binding properties,” mentioned Casim Sarkar, senior creator of the paper and a professor within the College of Minnesota Division of Biomedical Engineering.
“With multivalent medication, you possibly can, in precept, goal cells very particularly in a method that is not attainable with customary, monovalent medication, however there are numerous variables to think about of their design and far of the work within the area thus far has been accomplished by means of experimental trial and error,” Sarkar added. “Now, utilizing MVsim, we’re capable of make good predictions that can be utilized to extra rationally design such therapeutics.”
Many most cancers medication not solely bind to tumor cells but in addition to cells they don’t seem to be meant to focus on, which regularly creates undesirable negative effects for the affected person. By optimizing the specificity of multivalent interactions utilizing MVsim, researchers can design medication that extra particularly goal the cells in a tumor whereas minimizing binding to different cells within the physique.
One other instance is the SARS-CoV-2 virus. Scientists know that the virus is evolving to raised infect our cells and evade our immune techniques, however the molecular mechanisms behind how the virus does that is comparatively unknown. Utilizing their MVsim expertise, the College of Minnesota researchers had been capable of discover this course of extra in depth, uncovering the charges at which particular person binding domains inside the virus’s multivalent spike protein change between a cell-infecting state and an immune-evading state.
“We basically have a computational microscope that enables us to look underneath the hood and see what multivalent proteins such because the SARS-CoV-2 spike protein are doing at a molecular degree,” Sarkar defined. “This degree of molecular element is tough to seize with a bodily experiment. One of many actual powers of MVsim is that we cannot solely be taught extra about how these techniques work however we are able to additionally use this device to design new multivalent interactions for ailments like most cancers and COVID-19.”
The researchers have already recognized potential methods to restrict the infectivity of present and future SARS-CoV-2 variants, which they plan to check quickly.
Bence Bruncsics et al, MVsim is a toolset for quantifying and designing multivalent interactions, Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6
MVsim app: GitHub
Quotation:
Expertise that simulates complicated molecular interactions might result in higher therapies for most cancers and COVID-19 (2022, September 6)
retrieved 7 September 2022
from https://phys.org/information/2022-09-technology-simulates-complex-molecular-interactions.html
This doc is topic to copyright. Other than any truthful dealing for the aim of personal research or analysis, no
half could also be reproduced with out the written permission. The content material is supplied for info functions solely.